
Į³╚šŻ¼ųą─Ž┤¾īWŽµč┼Č■ßtį║ā║═»ßtīWųąą─č¬ę║─[┴÷īŻ┐Ų╩šų╬┴╦ę╗├¹ęįĪ░Ę┤Å═░l¤ßĪóļyų╬ąįžÜč¬Īó╔·ķL░lė²┬õ║¾Ī▒×ķ┼R┤▓▒Ē¼FĄ─2Üq┼«═»Ż¼ūĪį║║¾ĮY║Ž╣Ū╦Ķ╝Ü░¹īW┼cŠĆ┴Ż¾w╗∙ę“Öz£yį\öÓ×ķ║▒ęŖ▓ĪĪ¬Ī¬Ī░ŲżĀ¢▀dŠC║Žš„Ī▒ĪŻĮø▀^ų╬»¤Ż¼╗╝ā║─┐Ū░š¹¾wŪķørĘĆČ©Ż¼Ą½╚įėą▓╗═¼│╠Č╚žÜ謯¼ąĶķLŲ┌ė┌įōį║ā║┐Ųč¬ę║─[┴÷īŻ┐ŲķTį\ļSįL▓óČ©Ų┌ąą╝t╝Ü░¹▌öūó╝░┤·ųxš{╣Øų╬»¤ĪŻ
Į±─Ļ2į┬28╚š╩ŪĄ┌╩«░╦éĆć°ļH║▒ęŖ▓Ī╚šŻ¼Į±─ĻĄ─ų„Ņ}╩ŪĪ░▓╗ų╣║▒ęŖŻ©More than you can imagineŻ®Ī▒Ż¼ų╝į┌▀Mę╗▓Į╠ßĖ▀┤¾▒Ŗī”║▒ęŖ▓ĪĄ─ĻPūó║═šJų¬Ż¼═Ų▀M║▒ęŖ▓Ī┐ŲŲšą¹Į╠Ż¼┤┘▀M║▒ęŖ▀zé„▓ĪĄ─║Y▓ķĪóį\öÓ╝░ŅAĘ└Ż¼╠ßĖ▀╔·┤µ┘|┴┐ĪŻį┌▒ŖČÓ║▒ęŖ▓ĪųąŻ¼ŲżĀ¢▀dŠC║Žš„╚½Ū“ł¾Ėµ▓Ī└²▓╗│¼▀^150└²Ż¼ę“ŲõÅ═ļsąį║═ī”ā║═»Ą─ų┬├³═■├{Ż¼žĮąĶĖ³ČÓšJų¬┼cąąäėĪŻ
ŲżĀ¢▀dŠC║Žš„Ż©Pearson SyndromeŻ¼PSŻ®Ż¼╩Ūę╗ĘNė╔ŠĆ┴Ż¾wDNA╚▒╩¦ę²░lĄ─įŁ░ląįŠĆ┴Ż¾w▓ĪŻ¼░l▓Ī┬╩╝s×ķ░┘╚fĘųų«ę╗ĪŻŲõ░l▓ĪÖCųŲ╔µ╝░▀zé„╚▒Ž▌Īó┤·ųx╬╔üy╝░ĮM┐Ś«É┘|ąįĄ╚ČÓĘĮ├µę“╦žŻ¼╗╝ā║░lė²▀^│╠ųą▀@ĘNŠĆ┴Ż¾wDNA╚▒╩¦į┌Ė„ŽĄĮyĮM┐ŚųąĄ─└█Ęe▓╗═¼Ż¼Å─Č°ę²Ų▓╗═¼Ą─┼R┤▓▒Ē¼FĪŻ╦³Ž±ę╗Ą└ļ[ą╬Ą─┴č┐pŻ¼Ū─╚╗Ūųęu╗╝ā║Ą─č¬ę║ŽĄĮyĪóę╚Ž┘╝░ČÓéĆŲ„╣┘Ż¼ČÓöĄ╗╝ā║į┌│÷╔·║¾6éĆį┬ā╚░l▓ĪŻ¼▒╗ĘQ×ķĪ░╔·├³ūŅ│§Ą─╔·┤µ╠¶æĪ▒ĪŻ
ō■ŽżŻ¼╗╝ā║│§Ų┌│Ż▒Ē¼F×ķ▓╗═¼│╠Č╚Ą─žÜ謯¼┤¾ČÓ├µ╔½╔n░ūŻ╗░ķ╗“▓╗░ķėą░ū╝Ü░¹║═謹Ī░ÕĄ═Ž┬Ż¼Ėą╚Š╝░│÷謒LļUśOĖ▀Ż╗ ▓┐Ęų╗╝ā║░ķėąę╚Ž┘═ŌĘų├┌╣”─▄▓╗╚½Ż¼ų„ę¬▒Ē¼F×ķĖ╣×aĪóĀIB▓╗┴╝Ż¼ąĶę└┘ćŽ¹╗»├Ė╠µ┤·ų╬»¤Ż╗ų„ę¬▒Ē¼F×ķ╚ķ╦ßųąČŠŻ¼┐╔ī¦ų┬ČÓŲ„╣┘╣”─▄Ą─šŽĄKŻ╗╗╝ā║Ą─╔ĒĖ▀Īó¾wųžČÓĄ═ė┌═¼─Ļ²gČ╬Ųõ╦¹ā║═»ĪŻ┤╦═ŌŻ¼Ė╬┼KĪó─I┼KĪó╔±ĮøŽĄĮy┐╔─▄│÷¼F▀MąąąįĄ─ōpé¹Ż¼▓┐Ęų╗╝ā║║¾Ų┌╔§ų┴┐╔─▄░lš╣×ķĖ³ć└ųžĄ─Kearns-SayreŠC║Žš„Ż©č█╝Ī┬ķ▒įĪóęĢŠW─ż▓ĪūāŻ®ĪŻ
ė╔ė┌░YĀŅÅ═ļsŻ¼ŲżĀ¢▀dŠC║Žš„│Ż▒╗š`į\×ķŲš═©žÜč¬╗“Ėą╚ŠŻ¼┤_į\ę└┘ć╣Ū╦Ķ╝Ü░¹īWÖz▓ķ║═ŠĆ┴Ż¾wDNAĘų╬÷ĪŻ
─┐Ū░Ż¼┤_į\ĘĮĘ©═©▀^╗∙ę“Öz£yŻ¼╝┤═©▀^č¬ę║╗“╣Ū╦Ķśė▒ŠÖz£yŠĆ┴Ż¾wDNA┤¾Ų¼Č╬╚▒╩¦Ż╗╣Ū╦Ķ╗ŅÖzŻ¼╝┤┐╔ęįė^▓ņĄĮ╠žš„ąįĄ─╝tŽĄŪ░¾w╝Ü░¹Īó┴ŻŽĄŪ░¾w╝Ü░¹Ą─░¹┘|Ī░┐š┼▌╗»Ī▒ęį╝░Łhą╬ĶF┴Żėū╝Ü░¹ĪŻ
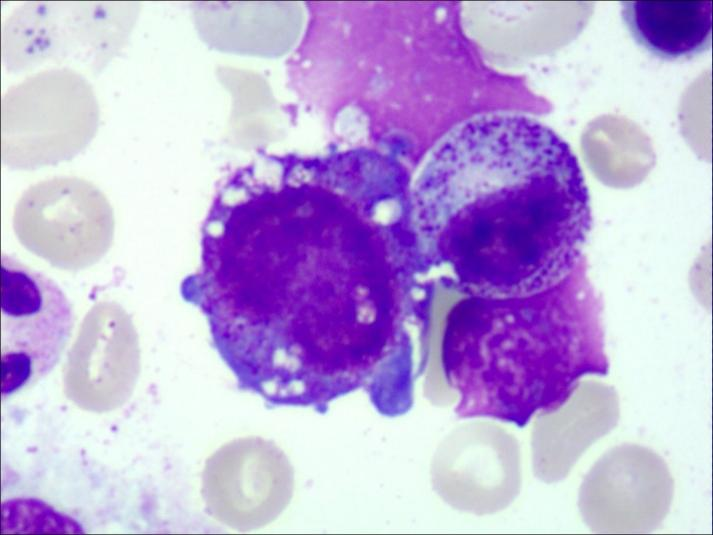
┴ŻŽĄŪ░¾w╝Ü░¹┐š┼▌╗»Ż©ųą─Ž┤¾īWŽµč┼Č■ßtį║ā║═»ßtīWųąą─ ╣®łDŻ®
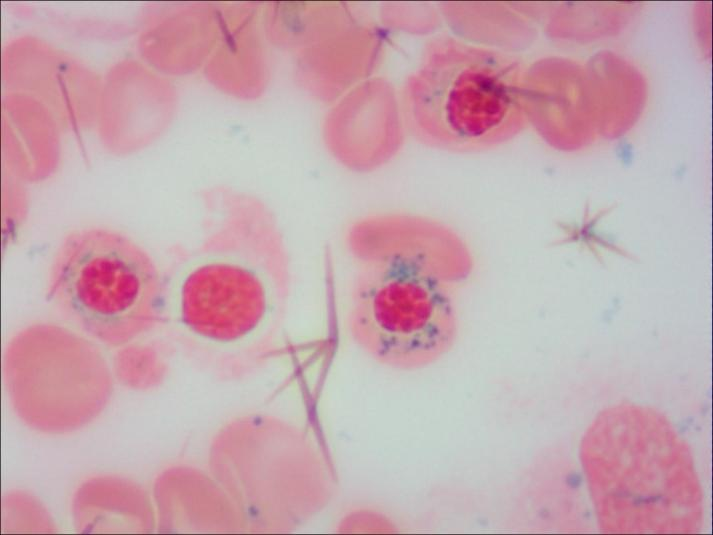
Łhą╬ĶF┴Żėū╝Ü░¹Ż©ųą─Ž┤¾īWŽµč┼Č■ßtį║ā║═»ßtīWųąą─ ╣®łDŻ®
─┐Ū░Ż¼ŲżĀ¢▀dŠC║Žš„╔ą¤oĖ∙ų╬ĘĮĘ©Ż¼─┐Ū░ęįų¦│ųų╬»¤×ķų„ĪŻ
ų„ę¬▓╔ė├▌öč¬║═ĶF“³║Žä®Ż¼ŠÅĮŌžÜ謯¼ŅAĘ└ĶF▀^▌dŻ╗ ę╚├Ė║═┤·ųxų¦│ųŻ¼Ė─╔ŲŽ¹╗»╬³╩šŻ¼ča│õŠS╔·╦ž╝░▌o├ĖęįĖ─╔ŲÖC¾w┤·ųxŻ╗ī”░Y╣▄└Ē┤·ųxå¢Ņ}Ż¼╚ń╝mš²╦ßųąČŠĪŻ
Į³─ĻüĒŻ¼┐ŲčąŅIė“ėŁüĒ═╗ŲŲŻ¼╚ń╗∙ę“»¤Ę©Ż¼ßśī”ŠĆ┴Ż¾wDNA╚▒Ž▌Ą─ą▐Å═╝╝ąg▀M╚ļ┼R┤▓įć“×ļAČ╬Ż╗ŠĆ┴Ż¾węŲų▓Ż¼╩ūéĆŠĆ┴Ż¾wį÷ÅŖ»¤Ę©Ż©MATŻ®┼R┤▓įć“×åóäėŻ©ClinicalTrials.gov ś╦ūRĘ¹Ż║NCT03384420Ż®Ż¼Įo╗╝š▀║═╝ę═źÄ¦üĒ┴╦ą┬Ą─ŽŻ═¹Ż╗┐Ųčąų·┴”╚½Ū“ŲżĀ¢▀dŠC║Žš„ŽÓĻPšō╬─▌^╔┘Ż¼ųąć°īWš▀š²ŅI┼▄╗∙ę“ų╬»¤ĪŻŻ©║·╦žįŲ ³SĀ¢╝诮
 ķL░┤Č■ŠS┤a
ķL░┤Č■ŠS┤aĻPūóŠ½▓╩ā╚╚▌





